
Mirror Orientation Selection (MOS)
Suppression subtractive hybridization (SSH) is one of the most powerful and popular methods for isolating differentially expressed transcripts. SSH is based on the suppression PCR effect that is mediated by long inverted terminal repeats attached to the ends of DNA fragments (Lukyanov et al., 1994; Gurskaya et al., 1996; Diachenko et al., 1996). SSH includes the following main steps: (i) subdivision of tester cDNA into two samples and ligation of these samples with two different suppression adapters; (ii) hybridization of tester with excess driver; and (iii) amplification of the tester cDNA molecules that are flanked only with different suppression adapters (this fraction contains the enriched and normalized target cDNA).
Two types of unwanted background clones representing non-differentially expressed transcripts can be generated by SSH. Unincorporated adapters can randomly anneal to non-target cDNA molecules during subtractive hybridization. After DNA elongation, such molecules can serve as a template for amplification and will be picked up in the final product, as will product generated by non-specific annealing of PCR primers.
Redundant cDNA molecules can evade elimination by hybridization with driver and will amplify in subsequent PCR steps, though this is extremely unlikely. In most cases just a single molecule of each redundant cDNA will exist among several thousand other cDNA molecules after the subtractive hybridization and will not amplify as a false positive. However, in the case of a very abundant sequence, sufficient excess can be present to allow amplification in subsequent steps.
Clones representing type I background can be identified by differential screening because they do not produce a differential signal. On the contrary, type II background clones show differential signals during screening with probes prepared from two reciprocal (forward and reverse) subtracted samples. Only Northern blot and RT-PCR analysis can demonstrate the equal abundance of such sequences in the initial mRNA samples. Therefore, elimination of this background is a difficult and time-consuming step in subtracted library analysis. As an alternative to screening with subtracted probes, it is possible to use the tester and driver cDNA as probes for differential screening, but in this case many clones representing rare transcripts give no signals.
Mirror Orientation Selection (MOS) effectively eliminates type II background.
Type II background molecules have only one orientation with respect to the two different flanking adapter sequences used in SSH, while truly differentially expressed target cDNA fragments are represented in both sequence orientations.
MOS dramatically decreases the portion of type II background in the subtracted samples (Rebrikov et al., 2000). The method is based on the rationale that after PCR, each species of background molecule has only one orientation relative to the adapter sequences. This directionality corresponds to the orientation of the progenitor molecule. On the contrary, the target cDNA fragments are involved in PCR amplification due to efficient enrichment in the SSH procedure. As a result, each specific sequence has many progenitors and is represented by both sequence orientations.
The strategy of MOS is depicted in the figure below. One adapter (adapter B) is removed by restriction endonuclease digestion, heat-denaturation and re-annealing of the SSH sample. Some of the new hybrids will bear adapter A at both termini as a result of hybridization between molecules with mirror orientation of adapters A and B. Because of their bidirectionality, these sequences may be defined as having come from the target cDNA fraction. Next, the 3' ends are filled in and PCR is performed with primers corresponding to adapter A. In this reaction, only molecules bearing adapter A at both termini will amplify, leading to target enrichment of the PCR product.
 | Schematic outline of MOS. |
|---|
MOS performance has been demonstrated in model and experimental systems (Rebrikov et al., 2000; Rebrikov et al., 2002). To create artificial tester samples, various amounts of bacteriophage φX174 DNA was used as a target for subtraction in human skeletal muscle double-stranded cDNA. The amount of φX174 DNA added was 0.01% or 0.001% of the human cDNA. The same cDNA without viral DNA was used as the driver. Both the tester and the driver were digested by HaeIII and used for SSH and subsequent MOS. After subtraction of the tester with 0.01% of the target, the pattern of bands corresponding to φX174 was clearly visible but a bright smear representing background cDNA was also present (lane 1; as compared to control φX174 DNA in lane 7). MOS application reduced the background as evidenced by the clarity of lane 2. The efficiency of MOS was even more obvious when we tested it against 0.001% target sequence. In this case the SSH product was an even smear with no apparent bands (lane 4) and obviously included a very low portion of the target DNA. However, after MOS the sample contained bright bands (lane 5) corresponding to φX174 fragments. In these model experiments we also tested the addition of excess driver (as described for the SSH method) in the hybridization mixture during MOS (lanes 3 and 6). It is apparent that the addition of driver did not enrich the target but did lead to the appearance of background bands, and is therefore not recommended.
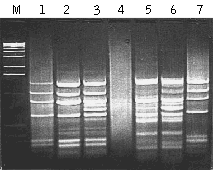 | Application of the MOS in a model subtraction.Gel electrophoresis of the PCR products: Lane M, 1 kb DNA ladder (Gibco BRL). Lanes 1-3, application of SSH and MOS to tester cDNA containing 0.01% φX174 DNA; lanes 4-6, application of SSH and MOS to tester cDNA containing 0.001% φX174 DNA. Lanes 1 and 3, samples after SSH. Lanes 2 and 5, samples after MOS. Lanes 3 and 6, samples after MOS with addition of excess driver. Lane 7, amplification product of φX174 DNA digested with HaeIII and ligated with adapters A and B. Small divergences in length of the fragments in SSH-generated, MOS-generated, and control φX174/HaeIII samples are due to differences in PCR primers used. |
|---|
References
- Diachenko L, Lau YF, Campbell AP, Chenchik A, Mogadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert D. (1996) Suppression Subtracive Hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc. Natl. Acad. Sci. USA 93(12): 6025-6030. / PMID:8650213
- Gurskaya NG, Diachenko L, Chenchik A, Siebert PD, Khaspekov GL, Lukyanov K, Vagner LL, Ermolaeva OD, Lukyanov S, Sverdlov ED. (1996) The Equalizing cDNA Subtraction Based on Selective Suppression of Polymerase Chain Reaction: Cloning of the Jurkat Cells' Transcripts Induced by Phytohemaglutinin and Phorbol 12-myristate 13-acetate. Anal. Biochem. 240(1): 90-97. / PMID:8811883
- Lukyanov SA, Gurskaya NG, Lukyanov KA, Tarabykin VS and Sverdlov ED (1994) Highly efficient subtractive hybridisation of cDNA. Russian Journal of Bioorganic Chemistry, 20 (6):701-704.
- Rebrikov DV, Britanova OV, Gurskaya NG, Lukyanov KA, Tarabykin VS and Lukyanov SA (2000) Mirror orientation selection (MOS) – a method for eliminating false positive clones from libraries generated by suppression subtractive hybridization. Nucleic Acid Research 28 (20): e90. / PMID:11024192
- Rebrikov DV, Bulina ME, Bogdanova EA, Wagner LL, Lukyanov SA. (2002) Complete genome sequence of a novel extrachromosomal virus-like element identified in planarian Girardia tigrina. BMC Genomics, 3(1):15. / PMID: 12065025